Supramolecular polymers
Noncovalent interactions provide a flexible means of engineering new chemical entities with tailored properties. Specific interactions between functionalized small molecules and polymer chains bearing complementary binding sites can be used to engineer supramolecular complexes that display mesomorphic structure. This has been exploited to develop a range of functional materials including photonic band gap polymers, ionic conductors and donor-acceptor semiconducting polymers. Additionally, the deliberate association of polymers with surfactants in engineered, synthetic materials is increasingly motivated by the possibility of combining the stimuli-responsive self-assembly and solubilizing properties of surfactants with the intrinsic solution properties of polymers, such as rheology modication and facile coating of interfaces. In solution, the hydrophobic nature of the surfactant compared to a hydrophilic polymer backbone leads to coil-globule transitions on decreasing solvent quality, surfactants cluster and force small length scale intrachain associations, causing a sharp reduction in the end-end chain distance, i.e collapse. These transitions qualitatively mimic the behavior of proteins in which there is an aggregation of hydrophobic side chains that occurs as a precursor to collapse and eventual folding. At higher concentrations, interchain associations drive supramolecular ordering, leading to larger characteristic length scales and in some cases to the formation of gels or networks. Overall, there is a compelling need to understand the physical chemistry, structure and dynamics of supramolecular polymers, both in solution and in the melt. Our work focuses on detailed examination of composition and temperature dependent molecular and supramolecular structures in solutions and melts. We quantitatively characterize the thermodynamics and kinetics of polymer-small molecule binding, elucidating the dependence on surfactant chemistry and environmental variables. We strive to formulate coherent frameworks describing structure-property relationships in the systems considered.
   |
Smectic demixing in hydrogen bonded LC polymers
Reversible hydrogen bonding between a rigid small molecule and a polymer having complementary sites for the surfactant can lead to phase separation and block copolymer-like morphology. Our studies are focused on the structure and dynamics of such complexes, with efforts paid to explicitly address the effect of the strength of the interaction between the polymer and surfactant. Above a critical stoichiometry, i.e. the ratio of ligand concentration to that of binding sites, these polymers may display liquid crystalline behavior in the melt state. The weak interaction between the ligands means that the binding equilibrium is dynamic, and so ligands are thought to change their sequence distribution along the polymer chain in response to changes in temperature, stoichiometry and the chemistry of the ligand. Characterizing this behavior is the central theme in our research.
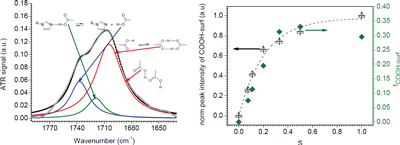
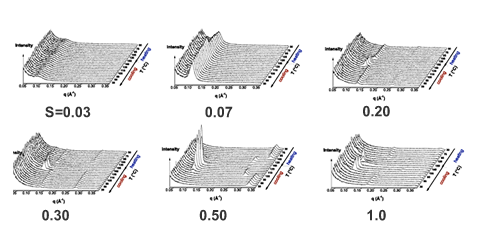
Over a limited range of temperatures and compositions, a supramolecular length scale emerges that is well beyond the upper limit imposed by a bilayer construct and thus cannot be accounted for within the conventional paradigm. Binding isotherms (Figure) reveal that the polymer has a limited capacity for the ligand with saturation occurring for S ≥ 0.33. These results suggest that the common assumption of homogeneously distributed tightly bound ligands with layer-like phase separation from the polymer backbone do not apply in this system over all compositions. The anomalous phase display is consistent with demixing between polymer rich and polymer poor domains due to the presence of excess unassociated mesogen, which can act as a solvent for the system.
 |
 |
Hairy-rod conjugated polymers and supramolecular semiconductors
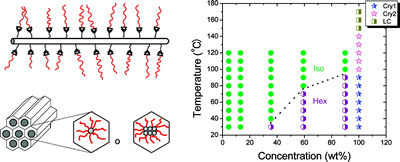
We study hydrogels formed by supramolecular hairy-rod conjugated polymers and their lyotropic liquid crystalline behavior. The system are ionic or other complexes between poly(thiophene) derivatives and various small molecules. For example, the Figure below shows data from negatively charged poly[2-(3-thienyl)ethyloxy-4-butylsulfonate)] backbones and cationic cetrimonium side chains. These hairy-rod supramolecules form stable isotropic solutions under dilute conditions with enhanced photoluminescence relative to the neat polymer itself. The system displays an isotropic-liquid crystalline transition on increasing concentration, resulting in the formation of a hexagonally ordered lyotropic mesophase in which the polymer chains are packed into hexagonally ordered rod-like assemblies. The mesophase is thermosensitive and can be isotropized by moderate heating. The use of oriented conjugated polymers in electro-optic devices can enhance device performance, for example through more efficient charge transport or decreased quenching of optical emissions. Lyotropic columnar phases represent a promising starting point for the production of highly aligned thin films by solution-based processing.
![]()
1.